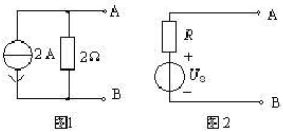
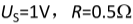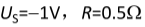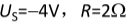题目内容
(请给出正确答案)
题目内容
(请给出正确答案)
[主观题]
图所示为三端网络,端子用a,d,e标记。已知R=1Ω,C=0.1F,L=0.5H,uab=sin10tV,udc=e-2tV,idc(0)=1A.求t>0时的ubc
图所示为三端网络,端子用a,d,e标记。已知R=1Ω,C=0.1F,L=0.5H,uab=sin10tV,udc=e-2tV,idc(0)=1A.求t>0时的ubc。

查看答案

 如果结果不匹配,请 联系老师 获取答案
如果结果不匹配,请 联系老师 获取答案

 更多“图所示为三端网络,端子用a,d,e标记。已知R=1Ω,C=0…”相关的问题
更多“图所示为三端网络,端子用a,d,e标记。已知R=1Ω,C=0…”相关的问题

1.jpg) (1)EcoRⅠ到HindⅢ位点间的片段有多大? (2)画出HhaⅠ和Sau3A的酶切位点。 (3)标记有32p的同样DNA片段,用HhaⅠ完全酶切,进行电泳,那么用EB染色或放射自显影,会分别得到什么样的图?
(1)EcoRⅠ到HindⅢ位点间的片段有多大? (2)画出HhaⅠ和Sau3A的酶切位点。 (3)标记有32p的同样DNA片段,用HhaⅠ完全酶切,进行电泳,那么用EB染色或放射自显影,会分别得到什么样的图?